글로벌 시장 진출의 초석이 되는 미국 식품의약국(FDA)의 문턱은 여전히 높다. 국내보다 훨씬 복잡하고 까다로운 규제 요건이 적용되면서 허가 과정에서 수년간 지연되거나 아예 무산되는 사례가 적지 않다.
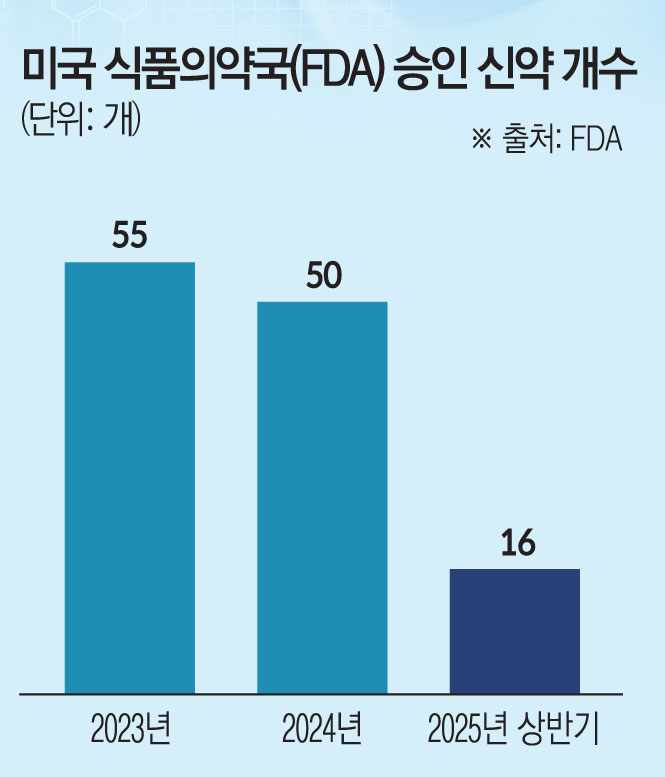
14일 제약 바이오업계에 따르면 FDA는 2023년에 55건, 2024년에는 50건의 신약을 승인했다. 그러나 올해 상반기에는 16건만 승인해 역대 최저 수준을 보였다. 최근 5년 상반기 평균(23개)과 비교하면 급감한 것을 확인할 수 있다.
국내 기업들도 FDA의 깐깐한 기준을 통과하는데 난항을 겪는다. HLB의 간암 신약 ‘리보세라닙’은 미국 FDA 문턱을 넘지 못하고 있다. 2022년 리보세라닙과 중국 항서제약의 캄렐리주맙 병용요법에 대한 간암 1차 치료제 글로벌 임상 3상 결과를 바탕으로 2023년 신약허가신청서(NDA)를 제출했지만, 2024년 5월 보완요구서한(CRL)을 받아 허가가 불발됐다. 항서제약 생산설비의 제조·품질관리(CMC) 문제였다. 이후 지적된 사항을 보완해 NDA를 다시 냈지만, 올해 3월 재차 CRL을 받았다. FDA가 최근 홈페이지에 공개한 CRL 원문에 따르면 항서제약에 대한 CMC 결함이 지적됐으나 구체적인 설명이나 내용은 비공개 처리했다. 비임상·임상 자료의 최신 안전성 업데이트와 복약 안내문, 최신 라벨링 및 번역본 제출도 요구했다.
GC녹십자의 면역글로불린 10% 제제 ‘알리글로’는 FDA 문턱을 넘는 데 무려 8년이 걸렸다. 2015년 면역글로불린 5% 제품에 대한 허가를 신청했지만, 2016년 11월과 2017년 제조공정 자료 보완으로 인해 허가가 미뤄졌다. 회사는 면역글로불린 10% 제품으로 미국에 진출하는 것으로 전략을 수정했고, 2021년 2월 품목허가 신청서를 제출했다. 하지만 코로나19로 인해 현장실사가 제때 이뤄지지 못했고, 2022년 CRL을 받았다. 2023년 상반기 현장실사를 다시 거친 뒤 비로소 같은 해 12월에 허가에 성공했다.
이렇게 FDA 허가 획득이 어려운 이유는 엄격한 안전성과 유효성 기준, 복잡하고 방대한 임상시험 절차, 까다로운 규제 환경 때문이다. 글로벌 빅파마들도 안전성 데이터 부족, 임상시험 결과 미흡, 제조시설 문제 등으로 고배를 마시는 경우가 흔하다. FDA는 서류 및 데이터 제출과 관련해 현존 규제기관 중 최고 수준의 정밀성과 일관성을 요구해 작은 오류만 있어도 CRL을 수령하게 된다.
국내 기업은 이에 더해 △영문 문서 작성 역량 부족 △글로벌 임상시험수탁기관(CRO) 네트워크 미비 △미국 현지 임상시험심사위원회(IRB) 대응 경험 부족 등 추가적인 걸림돌이 있다. 바이오업계 관계자는 “번역 품질이나 문서 완결성이 떨어지면 FDA는 곧바로 보완을 요구한다. 이 경우 허가 일정이 수년 단위로 늦어질 수 있다”라면서 “국내에서 임상·제조·규제 경험이 충분히 축적되지 않은 상태에서 무리하게 미국 진출을 추진하다가 좌절하는 경우가 많다”라고 말했다.
최근 FDA 내부의 변화로 부담은 더 커질 것으로 우려된다. FDA는 올해 3월 트럼프 행정부가 발표한 구조조정 계획에 따라 직원 약 3500명을 해고했다. 공식 발표에 따르면 심사관 인력은 유지했다고 하지만, 행정·지원 부서 축소로 문서 검토 속도는 느려질 수밖에 없다. 한 제약사 관계자는 “FDA 내부 혼란과 실사 지연이 겹치면서 허가 일정 예측이 더욱 어려워졌다”라고 토로했다.

![남아공에 졌는데도 한국 32강 진출 확률 94%⋯왜? [북중미 월드컵]](https://img.etoday.co.kr/crop/140/88/2351107.jpg)
![한국 축구대표팀, 이후 일정은? [북중미 월드컵]](https://img.etoday.co.kr/crop/140/88/2351028.jpg)
![‘안전자산’ 위상 잃은 금, 3년 강세장 끝났다…금리 인상 기조에 매력↓ [대체자산의 추락 ①]](https://img.etoday.co.kr/crop/140/88/2350093.jpg)
![10명 중 9명 "경제적 자유 달성해도 '일은 계속'" [데이터클립]](https://img.etoday.co.kr/crop/140/88/2351064.jpg)

![감독ㆍ축협ㆍ선수 모두 잘못⋯홍명보호 '전방위 직격' [북중미 월드컵]](https://img.etoday.co.kr/crop/140/88/2351048.jpg)















![10명 중 9명 "경제적 자유 달성해도 '일은 계속'" [데이터클립]](https://img.etoday.co.kr/crop/300/170/2351064.jpg)
![코스피 급등, 9000선 코앞 [포토로그]](https://img.etoday.co.kr/crop/300/190/2351057.jpg)